Lipids: Fats, Oils, Waxes, etc.
What Are Lipids?
All Lipids are hydrophobic: that’s the one property they have in common. This group of molecules includes fats and oils, waxes, phospholipids, steroids (like cholesterol), and some other related compounds.
Structure of Fatty Acids

One Fatty AcidThe “tail” of a fatty acid is a long hydrocarbon chain, making it hydrophobic. The “head” of the molecule is a carboxyl group which is hydrophilic. Fatty acids are the main component of soap, where their tails are soluble in oily dirt and their heads are soluble in water to emulsify and wash away the oily dirt. However, when the head end is attached to glycerol to form a fat, that whole molecule is hydrophobic.

Fatty AcidsThe terms saturated, mono-unsaturated, and poly-unsaturated refer to the number of hydrogens attached to the hydrocarbon tails of the fatty acids as compared to the number of double bonds between carbon atoms in the tail. Fats, which are mostly from animal sources, have all single bonds between the carbons in their fatty acid tails, thus all the carbons are also bonded to the maximum number of hydrogens possible. Since the fatty acids in these triglycerides contain the maximum possible amouunt of hydrogens, these would be calledsaturated fats. The hydrocarbon chains in these fatty acids are, thus, fairly straight and can pack closely together, making these fats solid at room temperature. Oils, mostly from plant sources, have some double bonds between some of the carbons in the hydrocarbon tail, causing bends or “kinks” in the shape of the molecules. Because some of the carbons share double bonds, they’re not bonded to as many hydrogens as they could if they weren’t double bonded to each other. Therefore these oils are called unsaturated fats. Because of the kinks in the hydrocarbon tails, unsaturated fats can’t pack as closely together, making them liquid at room temperature. Many people have heard that the unsaturated fats are “healthier” than the saturated ones. Hydrogenated vegetable oil (as in shortening and commercial peanut butters where a solid consistency is sought) started out as “good” unsaturated oil. However, this commercial product has had all the double bonds artificially broken and hydrogens artificially added (in a chemistry lab-type setting) to turn it into saturated fat that bears no resemblance to the original oil from which it came (so it will be solid at room temperature).

Cis and Trans BondsIn unsaturated fatty acids, there are two ways the pieces of the hydrocarbon tail can be arranged around a C=C double bond. In cis bonds, the two pieces of the carbon chain on either side of the double bond are either both “up” or both “down,” such that both are on the same side of the molecule. In trans bonds, the two pieces of the molecule are on opposite sides of the double bond, that is, one “up” and one “down” across from each other. Naturally-occurring unsaturated vegetable oils have almost all cis bonds, but using oil for frying causes some of the cis bonds to convert to trans bonds. If oil is used only once like when you fry an egg, only a few of the bonds do this so it’s not too bad. However, if oil is constantly reused, like in fast food French fry machines, more and more of the cis bonds are changed to trans until significant numbers of fatty acids with trans bonds build up. The reason this is of concern is that fatty acids with trans bonds are carcinogenic, or cancer-causing. The levels of trans fatty acids in highly-processed, lipid-containing products such as margarine are quite high, and the government now requires that the amounts of trans fatty acids in such products be listed on the labels.

Omega-3 and Omega-6 Fatty AcidsAnother set of fat-related terms that are “in the news” a lot, lately are “omega-3” and “omega-6” fatty acids. These terms both refer to fatty acids that have at least one unsaturated bond in their chains, and these terms describe where that bond is located. Starting from the end of the carbon chain (that does not contain the carboxyl group and is not bonded on to the glycerol), the carbon atoms before the first double bond are counted. If there are three carbons, it is an omega-3 fatty acid, and if there are are six carbons, it is an omega-6 fatty acid. We need a balanced amount of both in our diets, but the omega 3 fatty acids are not as common, thus harder to obtain, and so many people’s intake of these two types of fatty acids is way out of balance, including way too many omega-6 fats as compared to the amount of omega-3 fats in their diets. Flax seed and chia seed contain significant amounts of omega-3 fatty acids. Certain types of marine algae manufacture lots of omega-3 fatty acids, and those are incorporated into the tissues of fish which eat those algae. Because of that, many people take fish-oil capsules to increase the amount of omega-3 fats in their diets, but especially for vegetarians, consuming algae-derived omega-3 fats is another option.
Structure of Fats and Oils

Glycerol
TriglycerideFats and oils are made from two kinds of molecules: glycerol (a type of alcohol with a hydroxyl group on each of its three carbons) and threefatty acids joined by dehydration synthesis. Since there are three fatty acids attached, these are known as triglycerides. “Bread” and pastries from a “bread factory” often contain mono- and diglycerides as “dough conditioners.” Can you figure out what these molecules would look like? The main distinction between fats and oils is whether they’re solid or liquid at room temperature, and this, as we’ll soon see, is based on differences in the structures of the fatty acids they contain.
The fatty acids that make up the fats and oils in our diets and our bodies may also be lumped into groups based upon the number of carbon atoms in their chains. Interestingly, most of the fatty acids in living organisms have an even number of carbons (2, 4, 6,…). Fatty acids with less than 6 carbons in their chains may collectively be called short-chain fatty acids, but usually these are just called carboxylic acids and are not referred to as “fatty” acids. Those with 6 to 12 carbons are medium-chain fatty acids, those with 14 to 22 carbons are long-chain fatty acids, and those with over 22 carbons are thevery-long-chain fatty acids. Since, as mentioned above, fats and oils contain three fatty acids and are called triglycerides, those which contain primarily medium-chain fatty acids are referred to as medium-chain triglycerides (MCTs), while those which contain primarily long-chain fatty acids are referred to as long-chain triglycerides (LCTs). Most of the fats and oils in our diets and that we’re used to hearing about in the news are LCTs.
| Chain Length | # of Carbons | Name | Formula | Notes |
|---|---|---|---|---|
| The first 11 saturated fatty acids are: | ||||
| short-chain | C2 | acetic acid | CH3COOH | aceto = vinegar |
| C4 | butryic acid | CH3(CH2)2COOH | butyr = butter | |
| medium-chain | C6 | caproic acid | CH3(CH2)4COOH | capri = goat |
| C8 | caprylic acid | CH3(CH2)6COOH | ||
| C10 | capric acid | CH3(CH2)8COOH | ||
| C12 | lauric acid | CH3(CH2)10COOH | lauri = laurel | |
| long-chain | C14 | myristic acid | CH3(CH2)12COOH | myrist = anoint, ointment |
| C16 | palmitic | CH3(CH2)14COOH | ||
| C18 | stearic acid | CH3(CH2)16COOH | stear = fat, suet, tallow | |
| C20 | arachidic or eicosanoic acid | CH3(CH2)18COOH | arachis = a leguminous plant, present in peanut oil | |
| C22 | behenic or docosanoic acid | CH3(CH2)20COOH | present peanut & canola oils | |
| A few of the more important unsaturated fatty acids include: | ||||
| EFA = essential fatty acid, PUFA = polyunsaturated fatty acid, MUFA = monounsaturated fatty acid. (CH2)7, for example, means repeat CH2 seven times in a row, so CH2CH2CH2CH2CH2CH2CH2. To determine the omega number, count the number of carbons in from the left end of each of the following molecules until the first double bond. Do not start counting from the carboxyl group. |
||||
| C18 | oleic acid | CH3(CH2)7CH=CH(CH2)7COOH | omega-9, MUFA, present in olive & sesame oils, oleo = olive, olive oil | |
| C18 | linoleic acid | CH3(CH2)4CH=CHCH2CH=CH(CH2)7COOH | EFA, omega-6, PUFA | |
| C18 | linolenic or alpha linolenic acid (ALA) | CH3CH2CH=CHCH2CH=CHCH2CH=CH(CH2)7COOH | EFA, omega-3, PUFA, present in flaxseed oil | |
| C20 | arachidonic acid (ARA) | CH3(CH2)4(CH=CHCH2)4(CH2)2COOH | EFA, omega-6, PUFA | |
| C20 | eicosapentaenoic or timnodonic acid (EPA) | CH3(CH2CH=CH)5(CH2)3COOH | omega-3, PUFA, present in algae/fish oil | |
| C22 | docosahexaenoic or cervonic acid (DHA) | CH3(CH2CH=CH)6(CH2)2COOH | omega-3, PUFA, present in algae/fish oil | |
Notice that, while these unsaturated fatty acids contain the same number of carbons as some of the saturated fatty acids, they are chemically different and have different names.
The medium-chain triglycerides (MCTs) play some very interesting roles in our bodies. MCTs are handled quite differently by our bodies. Our digestive tract doesn’t need to digest them or even use bile to emulsify them, but they are directly absorbed into our blood and sent to the liver where some of them are converted into ketones. Both the MCTs themselves and the ketones formed from them are directly used by our brains and our muscles as alternate fuel/energy sources in place of glucose. In general, MCTs are not stored as body fat, so unless a person would consume way more than necessary, they do not contribute to weight gain. However, consumption of “too much” is unlikely because eating more at once than one’s body is used to tends to cause diarrhea. Even though they are saturated fats, they are “heart-healthy,” and the heart actually beats more effeciently using them as fuel in place of glucose.
Think what life must have been like back in “cave-man days” — people back then weren’t assured of three sumptous, regularly-scheduled meals a day, but rather, if they hadn’t killed a buffalo or a gazelle in several days, they might have nothing to eat for a couple days. If glucose (sugar) was the only fuel their brains and muscles could use to keep going, they would have, long-ago, starved to death, and we wouldn’t be here today. Rather, being able to use MCTs as an energy source enabled them to keep going and survive. Even today, mothers’ milk is very high in MCTs (10 to 17% of its fat), newborns’ brains use MCTs and their metabolic derivatives for as much as 25% of their energy requirements, and now, every infant formula on the market these days contains MCTs.
In terms of our brains’ abilities to use glucose as a fuel, it turns out that the insulin produced by our pancreas that helps transport sugar into all our other body cells (so it can be used as an energy source) cannot cross the blood-brain barrier, but rather, our brains make their own insulin which is used to help transport glucose into the neurons (nerve cells, brain cells) so they can use it for fuel. In terms of pancreatic insulin, you’ve probably heard of type I and type II diabetes, in which either the person’s pancreas isn’t making enough insulin or else the cells that need to take the sugar out of the blood have “broken” insulin receptors, so even if the person’s pancreas is making enough insulin, that “message” to take sugar out of the blood never gets received by the cells. While we tend to think about that in terms of all the sugar that’s staying in the blood, think for a minute what that means in terms of the cells that aren’t getting the sugar inside when they need it.
Now, instead of the rest of the body, think of all this in terms of the brain cells. Don’t think in terms of blood or in terms of fluid within the brain, but think in terms of the actual brain cells, the neurons. What if a person’s neurons were insulin resistant? That would mean that sugar couldn’t get into the neurons to be used as energy to keep the neurons functioning. The neurons would end up, essentially, starving to death. Does that sound far-fetched or impossible? It turns out, based on current research, that can and does happen. Alzheimer’s is now being referred to as type III diabetes. It has been observed that many people who have been diagnosed with Alzheimer’s or other similar neurological diseases were “sugar junkies” for years before their diagnosis. While these people weren’t showing Alzheimer’s signs and symptoms, yet, their neurons were starving, even then, and were sending out messages “begging” for more sugar. Unfortunately, because those neurons were unable to get all that sugar inside themselves, it didn’t do any good, and they began to die off. When enough neurons had died, and thus, the person’s brain had “shrunk” enough for the deficit to be noticeable, then the person was diagnosed with Alzheimer’s or one of the other, closely-related neurological diseases.
MCTs are also being used to treat some types of cancer. Some very aggressive, rapidly-growing cancers can only use glucose as an energy source. Thus it has been found that, if people with those types of cancer are put on a special diet that is very low in sugar, but has adequate MCTs, the person’s body can use the MCTs for fuel, while the cancer starves.
We need fats in our bodies and in our diet. Animals in general use fat for energy storage because fat stores 9 KCal/g of energy. Plants, which don’t move around, can afford to store food for energy in a less compact but more easily accessible form, so they use starch (a carbohydrate, NOT A LIPID) for energy storage. Carbohydrates and proteins store only 4 KCal/g of energy, so fat stores over twice as much energy/gram as fat. By the way, this is also related to the idea behind some of the high-carbohydrate weight loss diets. The human body burns carbohydrates and fats for fuel in a given proportion to each other. The theory behind these diets is that if they supply carbohydrates but not fats, then it is hoped that the fat needed to balance with the sugar will be taken from the dieter’s body stores. Fat is also is used in our bodies to a) cushion vital organs like the kidneys and b) serve as insulation, especially just beneath the skin.
Phospholipids

LecithinPhospholipids are made from glycerol, two fatty acids, and (in place of the third fatty acid) a phosphate group with some other molecule attached to its other end. The hydrocarbon tails of the fatty acids are still hydrophobic, but the phosphate group end of the molecule is hydrophilic because of the oxygens with all of their pairs of unshared electrons. This means that phospholipids are soluble in both water and oil.
 An emulsifying agent is a substance which is soluble in both oil and water, thus enabling the two to mix. A “famous” phospholipid is lecithin which is found in egg yolk and soybeans. Egg yolk is mostly water but has a lot of lipids, especially cholesterol, which are needed by the developing chick. Lecithin is used to emulsify the lipids and hold them in the water as an emulsion. Lecithin is the basis of the classic emulsion known as mayonnaise. For more information on mayonnaise, see the Biol 1081L Mayonnaise Web page.
An emulsifying agent is a substance which is soluble in both oil and water, thus enabling the two to mix. A “famous” phospholipid is lecithin which is found in egg yolk and soybeans. Egg yolk is mostly water but has a lot of lipids, especially cholesterol, which are needed by the developing chick. Lecithin is used to emulsify the lipids and hold them in the water as an emulsion. Lecithin is the basis of the classic emulsion known as mayonnaise. For more information on mayonnaise, see the Biol 1081L Mayonnaise Web page.

Phospholipid BilayerOur cell membranes are made mostly of phospholipids arranged in a double layer with the tails from both layers “inside” (facing toward each other) and the heads facing “out” (toward the watery environment) on both surfaces.
Steroids

CholesterolThe general structure of cholesterol consists of two six-membered rings side-by-side and sharing one side in common, a third six-membered ring off the top corner of the right ring, and a five-membered ring attached to the right side of that. The central core of this molecule, consisting of four fused rings, is shared by allsteroids, including estrogen (estradiol), progesterone, corticosteroids such as cortisol (cortisone), aldosterone, testosterone, and Vitamin D. In the various types of steroids, various other groups/molecules are attached around the edges. Know how to draw the four rings that make up the central structure.
Cholesterol is not a “bad guy!” Our bodies make about 2 g of cholesterol per day, and that makes up about 85% of blood cholesterol, while only about 15% comes from dietary sources. Cholesterol is the precursor to our sex hormones and Vitamin D. Vitamin D is formed by the action of UV light in sunlight on cholesterol molecules that have “risen” to near the surface of the skin. At least one source I read suggested that people not shower immediately after being in the sun, but wait at least ½ hour for the new Vitamin D to be absorbed deeper into the skin. Our cell membranes contain a lot of cholesterol (in between the phospholipids) to help keep them “fluid” even when our cells are exposed to cooler temperatures.
Many people have heard the claims that egg yolk contains too much cholesterol, thus should not be eaten. An interesting study was done at Purdue University back in 1977 to test this. Men in one group each ate an egg a day, while men in another group were not allowed to eat eggs. Each of these groups was further subdivided such that half the men got “lots” of exercise while the other half were “couch potatoes.” The results of this experiment showed no significant difference in blood cholesterol levels between egg-eaters and non-egg-eaters while there was a very significant difference between the men who got exercise and those who didn’t.
Lipoproteins are clusters of proteins and lipids all tangled up together. These act as a means of carrying lipids, including cholesterol, around in our blood. There are two main categories of lipoproteins distinguished by how compact/dense they are. LDL or low density lipoprotein is the “bad guy,” being associated with deposition of “cholesterol” on the walls of someone’s arteries. HDL or high density lipoprotein is the “good guy,” being associated with carrying “cholesterol” out of the blood system, and is more dense/more compact than LDL.
References:
Borror, Donald J. 1960. Dictionary of Root Words and Combining Forms. Mayfield Publ. Co.
Campbell, Neil A., Lawrence G. Mitchell, Jane B. Reece. 1999. Biology, 5th Ed. Benjamin/Cummings Publ. Co., Inc. Menlo Park, CA. (plus earlier editions)
Campbell, Neil A., Lawrence G. Mitchell, Jane B. Reece. 1999. Biology: Concepts and Connections, 3rd Ed. Benjamin/Cummings Publ. Co., Inc. Menlo Park, CA. (plus earlier editions)
Lappé, Francis Moore. 1982. Diet for a Small Planet, 10th Anniversary Ed. Ballantine Books. New York.
Lappé, Francis Moore. 1991. Diet for a Small Planet, 20th Anniversary Ed. Ballantine Books. New York.
Marchuk, William N. 1992. A Life Science Lexicon. Wm. C. Brown Publishers, Dubuque, IA.
Sienko, Michell J. and Robert A. Plane. 1966. Chemistry: Principles and Properties. McGraw-Hill Book Co., NY. (and other chemistry texts and handbooks)



 Fatty acids made up of ten or more carbon atoms are nearly insoluble in water, and because of their lower density, float on the surface when mixed with water. Unlike paraffin or other alkanes, which tend to puddle on the waters surface, these fatty acids spread evenly over an extended water surface, eventually forming a monomolecular layer in which the polar carboxyl groups are hydrogen bonded at the water interface, and the hydrocarbon chains are aligned together away from the water. This behavior is illustrated in the diagram on the right. Substances that accumulate at water surfaces and change the surface properties are called surfactants.
Fatty acids made up of ten or more carbon atoms are nearly insoluble in water, and because of their lower density, float on the surface when mixed with water. Unlike paraffin or other alkanes, which tend to puddle on the waters surface, these fatty acids spread evenly over an extended water surface, eventually forming a monomolecular layer in which the polar carboxyl groups are hydrogen bonded at the water interface, and the hydrocarbon chains are aligned together away from the water. This behavior is illustrated in the diagram on the right. Substances that accumulate at water surfaces and change the surface properties are called surfactants.

















 CnH2nOn + n O2
CnH2nOn + n O2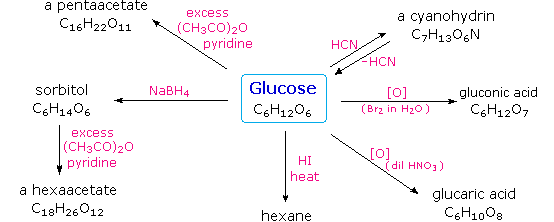
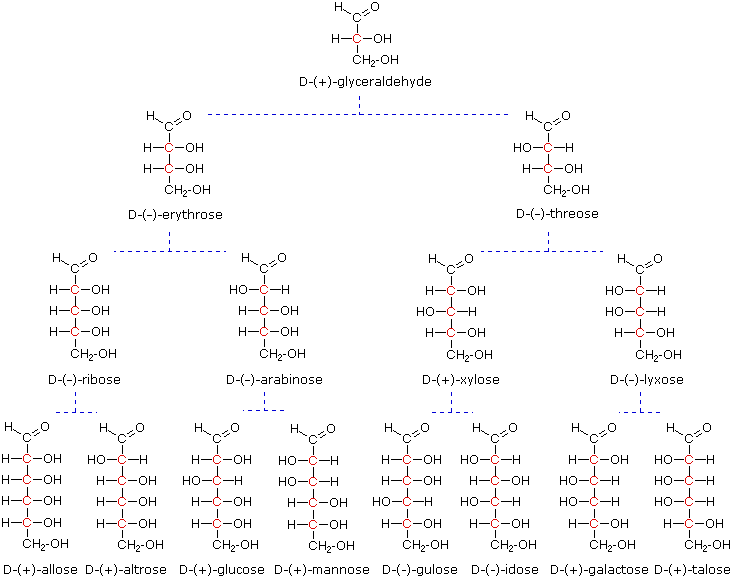
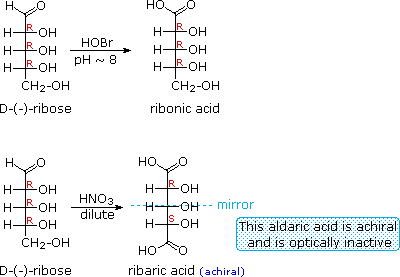
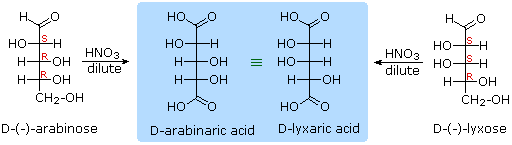
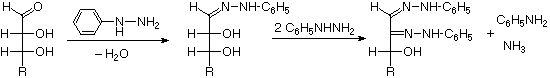
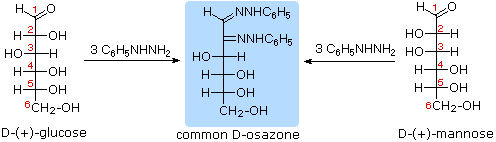

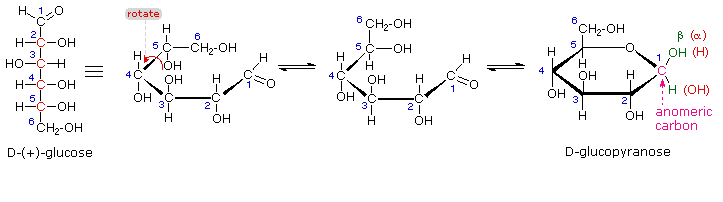
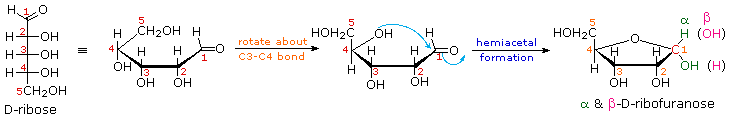
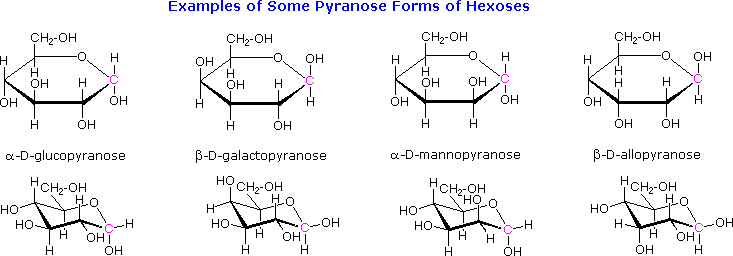
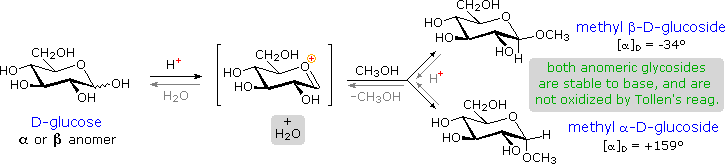
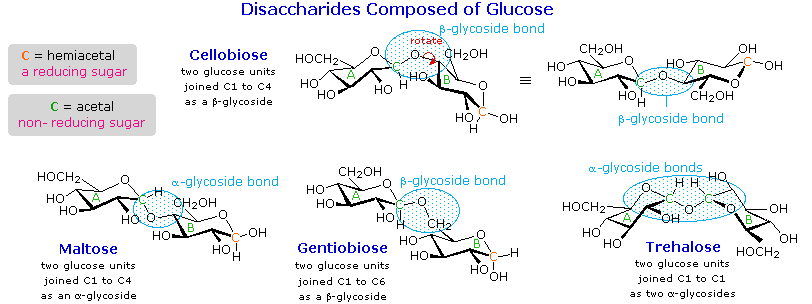
 As noted above, the preferred structural form of many monosaccharides may be that of a cyclic hemiacetal. Five and six-membered rings are favored over other ring sizes because of their low
As noted above, the preferred structural form of many monosaccharides may be that of a cyclic hemiacetal. Five and six-membered rings are favored over other ring sizes because of their low